| |  | |
-
Complexes with Redox-Active Ligands: Synthesis, Structure, and Electrochemical and Photophysical Behavior of the Ru(II) Complex with TTF-Annulated Phenanthroline
L.K. Keniley, N. Dupont, L. Ray, J. Ding, K. Kovnir, J.M. Hoyt, A. Hauser and M. Shatruk
Inorganic Chemistry, 52 (14) (2013), p8040-8052


DOI:10.1021/ic4006949 | unige:28963 | Abstract | Article HTML | Article PDF
|
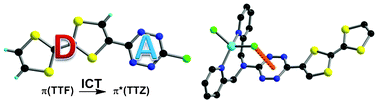
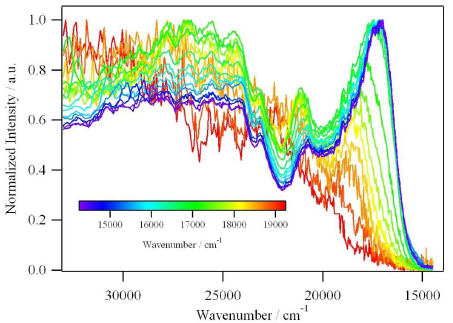
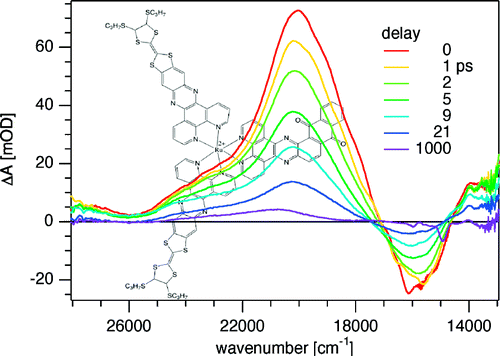
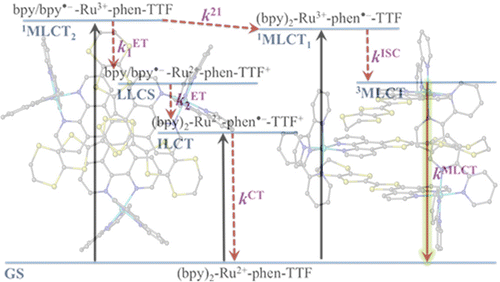
Ru(II) complexes with chelating ligands, 4â²,5â²-ethylenedithiotetrathiafulvenyl[4,5-f][1,10]phenanthroline (L1), 1,3-dithiole-2-thiono[4,5-f][1,10]phenanthroline (L2), and 1,3-dithiole-2-ono[4,5-f][1,10]phenanthroline (L3), have been prepared and their structural, electrochemical, and photophysical properties investigated. Density functional theory (DFT) calculations indicate that the highest occupied molecular orbital of [Ru(bpy)2(L1)](PF6)2 (1) is located on the tetrathiafulvalene (TTF) subunit and appears â0.6 eV above the three Ru-centered d orbitals. In agreement with this finding, 1 exhibits three reversible oxidations: the two at lower potentials take place on the TTF subunit, and the one at higher potential is due to the Ru3+/Ru2+ redox couple. Complexes [Ru(bpy)2(L2)](PF6)2 (2) and [Ru(bpy)2(L3)](PF6)2 (3) exhibit only the Ru3+/Ru2+-related oxidation. The optical absorption spectra of all complexes reveal a characteristic metal-to-ligand charge transfer (MLCT) band centered around 450 nm. In addition, in the spectrum of 1 the MLCT band is augmented by a low-energy tail that extends beyond 500 nm and is attributed to the intraligand charge transfer (ILCT) transition of L1, according to time-dependent DFT calculations. The substantial decrease in the luminescence quantum yield of 1 compared to those of 2 and 3 is attributed to the reductive quenching of the emissive state via electron transfer from the TTF subunit to the Ru3+ center, thus allowing nonradiative relaxation to the ground state through the lower-lying ILCT state. In the presence of O2, complex 1 undergoes a photoinduced oxidative cleavage of the central CâC bond of the TTF fragment, resulting in complete transformation to 3. This photodegradation process was studied with 13C NMR and optical absorption spectroscopy. |